Folding and Insertion of Membrane Proteins.
Folding of β-barrel Membrane Proteins
Integral membrane proteins are normally very hydrophobic and insoluble in water. It is therefore difficult to design model systems to examine the principles of membrane protein folding and insertion in vitro. These model systems are nonetheless necessary to better understand the molecular basis of membrane protein folding by employing spectroscopic techniques. Soluble proteins are folded such that the hydrophobic residues are oriented towards the interior of the protein while hydrophilic residues are mostly located at the surface of the protein. Membrane proteins, in contrast, have a more complex surface structure. The outer surface of the transmembrane domain has to be hydrophobic to allow a stable integration of the protein within the hydrophobic core of the lipid bilayer. Other domains of membrane proteins, which are exposed to the aqueous space, have a surface polarity comparable to that of soluble proteins. The interior of a membrane protein can also be very polar as is the case for example for channel proteins. It is the interaction with the lipid bilayer that stabilzes the native fold of the transmembrane domain. In absence of supramolecular amphiphilic structures (lipid bilayers or detergent micelles) membrane proteins usually misfold and aggregate. In order to understand the folding events of a membrane protein, it has to be transformed into a non-native state from which it can be refolded. For example bacteriorhodopsin has been partially denatured in SDS micelles and regained most of its activity when mixed with micelles of DMPC and CHAPS in the presence of retinal. It is therefore a popular model to study membrane protein folding. However, Bacteriorhodopsin does not completely unfold in SDS and still possesses significant content of α-helical structure. Furthermore, most refolding experiments were so far carried out into mixed micelles of DMPC and CHAPS. Fragments of Bacteriorhodopsin have also been folded in the presence of DMPC vesicles. To study the folding of membrane proteins in detail, two experimental problems must be addressed. First, denaturation conditions must be found that result in up to 100% loss of tertiary and secondary structure. Second, it must be possible to completely refold the protein upon removal of the denaturant. Refolding could either take place into detergent micelles or, preferably, lipid bilayers.
Some progress has been made in denaturing and solubilizing membrane proteins. It was shown that integral membrane proteins of the β-barrel type, for instance porin of the E.coli outer membrane, can be fully denatured in 8 M urea These proteins will spontaneously insert and refold when the urea is strongly diluted by mixing with a large volume of urea-free buffer containing lipid vesicles in the liquid-crystalline phase (Surrey & Jähnig, 1992). Denaturation and refolding of these proteins can be carried out in absence of detergent. Based on these observations, it is possible to study folding and insertion of β-barrel proteins extensively.
Recent studies (Kleinschmidt & Tamm, 1996, 1999; Kleinschmidt et al., 1999a,b), performed at the University of Virginia, were focussed on the in vitro refolding and membrane insertion of E. coli Outer Membrane Protein A (OmpA). This protein is predicted to form an eight stranded β-barrel in the outer membrane of E. coli with a periplasmic part of approximately 150 residues.
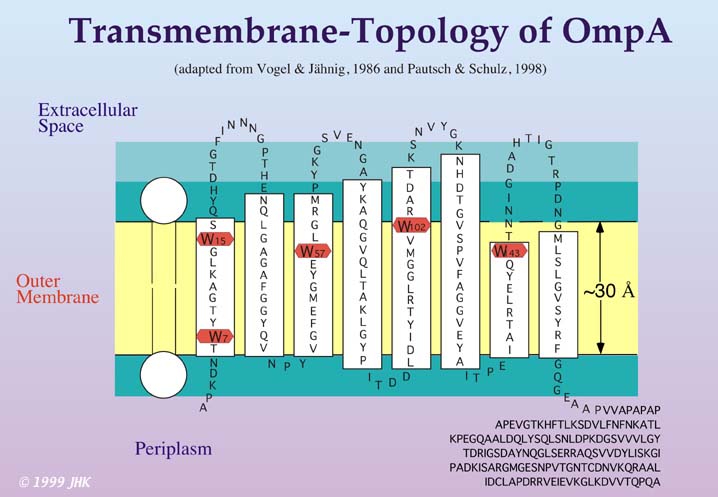
Evidence for this structure was provided previously by various mutagenesis studies as well as spectroscopic techniques investigating secondary structure, such as CD, FT-IR and Raman spectroscopy. The predicted transmembrane domain (residues 1-171) was recently crystallized by Pautsch and Schulz (Pautsch, A., Schulz, G. E., 1998, Nature Struct. Biol. 5, 1013-1017) and the x-ray crystal structure can be obtained from the Brookhaven Protein Database, entry 1BXW.
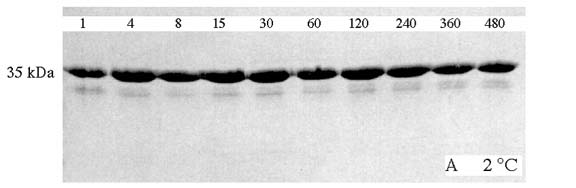
OmpA was also shown to be heat-modifiable. Purified OmpA in detergent would migrate on SDS-gels at apparent molecular weights of 35 or 30 kDa, depending on whether it was heated in SDS or not prior to loading the gel. In my studies, these properties of OmpA were utilized to investigate in vitro membrane protein insertion and folding into lipid bilayers using fluorescence spectroscopy and simple incubation experiments, which monitor how much folded OmpA (migrating at 30 kDa on SDS-gels) is developed as a function of incubation time after mixing the denatured protein with preformed vesicles. A combination of both techniques was used to study basic aspects of OmpA insertion and refolding. The Figure below shows refolding of OmpA at 40 °C. In this experiment, OmpA was not boiled in SDS, before the samples were loaded onto the gel. OmpA was first denatured in 8 M urea, which results in a complete random coil structure of the protein very similar to the heat denaturing in SDS. Such unfolded OmpA migrates at 35 kDa (upper bands). The denatured OmpA in 8 M urea is then strongly diluted in presence of preformed small unilamellar vesicles of dioleoyl-phoshatidylcholine (DOPC) at a 400:1 lipid/protein ratio. Incubation is stopped by addition of SDS treatment buffer at different times (as indicated in minutes on top of each lane). The gel indicates that approximately half of the protein folds within 30 min after initiation of the process.

If this experiment is carried out at 2 °C, OmpA will not fold completely as demonstrated in the gel below.
The kinetics of this process were studied by fluorescence spectroscopy at the different temperatures. At 2 °C the intrinsic fluorescence of the protein increased in the presence of lipid bilayers at a wavelength of 330 nm. The fluorescence intensity maximum shifted during folding from 354 nm in 8 M urea to 334 nm in presence of DOPC. This shift was more pronounced than the shift observed for the hydrophobic collapse in buffer without DOPC vesicles (fluorescence maximum at a wavelength of 343 nm), which indicates OmpA interaction with the lipid bilayer at this temperature. The time course of the fluorescence of OmpA for folding at 2 °C is shown in the figure below.
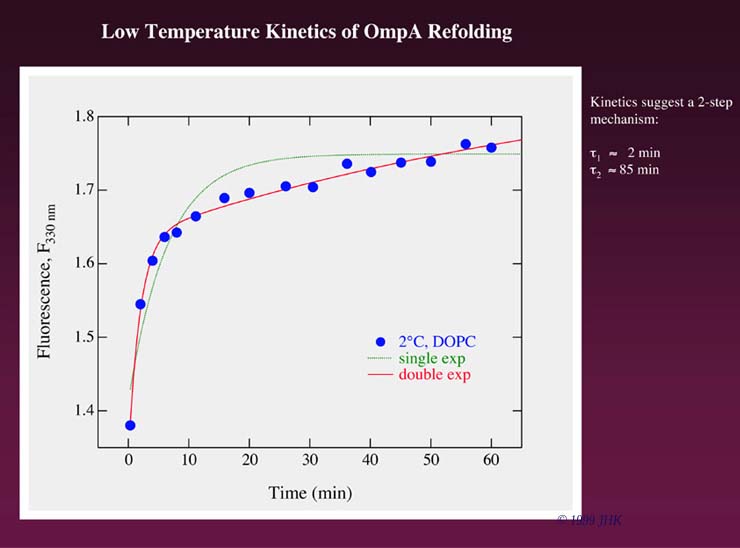
Fluorescence kinetics of OmpA refolding at 2 °C are best described by a double exponential time course. Since OmpA does not fully fold at 2 °C (see gel above) the fluorescence indicates that there must be two membrane bound states of OmpA that precede folding to the native state. One might argue that the first, fast step observed in the fluorescence kinetics represents the formation of hydrophobically collapsed OmpA. However, this collapse will occur much faster, on a millisecond rather than on a second or minute time scale, as an immediate result of urea dilution. That the fast step indeed represents a first absorption of OmpA to the lipid bilayer surface is immediately clear when the wavelength shifts of the fluorescence intensity maximum in presence and in absence of lipid membranes are compared as shown in the figure below.
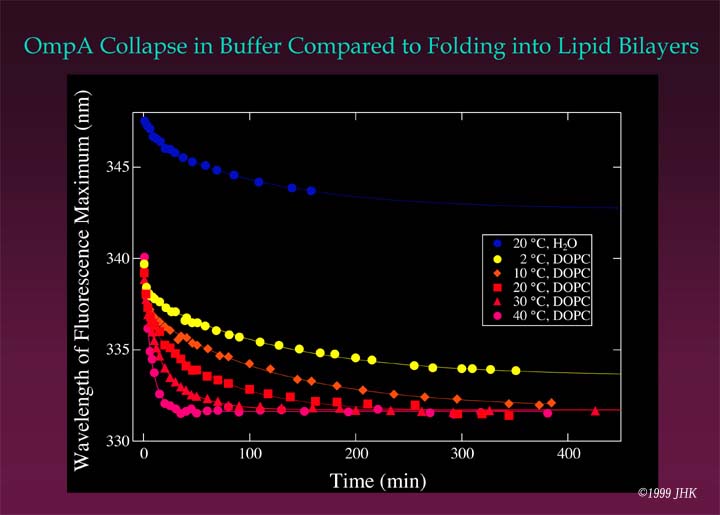
In presence of lipid bilayers, fluorescence changes much more quickly to a wavelength of 340 nm for the fluorescence maximum within the first 20 s after initiation of folding. In absence of lipid bilayers the wavelength shift of the intensity maximum is from 354 nm in 8 M urea to ~348 nm in buffer within the first 20 s. The later phase of refolding in buffer leads to a misfolded/aggregated state of OmpA. In lipid bilayers, folding completes via two kinetically distinct membrane associated intermediates (see cartoon below).
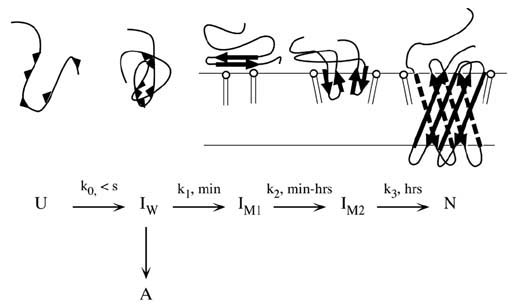
Conclusion: It was found, that at least 3 folding intermediates exist before unfolded OmpA is completely refolded. (Kleinschmidt, J. H. & Tamm, L. K., 1996, Biochemistry 35, 12993-13000). This model was subsequently extended using a new method, Time Resolved Distance Determinations by Fluorescence Quenching (TDFQ).
Back to homepage
(c) 1998,1999, Jörg Kleinschmidt, all rights reserved.